实际上,predict 唯一需要的是 newdata 中的列名与公式中使用的列名完全匹配。而且,你必须为每个预测变量提供一个值。以下是一些示例数据。
set.seed(16)
data <- data.frame(
mating=sample(0:1, 200, replace=T),
pop=sample(letters[1:4], 200, replace=T),
behv = scale(rpois(200,10)),
condition = scale(rnorm(200,5))
)
data1<-data[1:150,]
data2<-data[51:200,-1]
接下来,将使用data1拟合模型并预测结果到data2中。
model<-glm ( mating ~ behv * pop +
I(behv^2) * pop + condition,
data=data1,
family=binomial(logit))
predict(model, newdata=data2, type="response")
使用
type="response" 将为您提供预测概率。
现在,要进行预测,您不必使用来自完全相同的
data.frame 的子集。您可以创建一个新的数据框来调查特定范围的值(只需确保列名匹配即可)。因此,为了探索
behv*pop2(或在我的示例数据中的
behv*popb),我可以创建像这样的一个数据框。
popbbehv<-data.frame(
pop="b",
behv=seq(from=min(data$behv), to=max(data$behv), length.out=100),
condition = mean(data$condition)
)
在这里,我修正了
pop="b",所以我只关注
pop,由于我必须提供
condition,因此我将其修正为原始数据的平均值。(我本可以输入0,因为数据已经居中和缩放了。)现在我指定了一段我感兴趣的
behv值的范围。在这里,我只取了原始数据的范围,并将其分成100个区域。这将给我足够多的点来绘制图形。所以我再次使用
predict来得到。
popbbehvpred<-predict(model, newdata=popbbehv, type="response")
然后我可以使用某种工具来绘制它。
plot(popbbehvpred~behv, popbbehv, type="l")
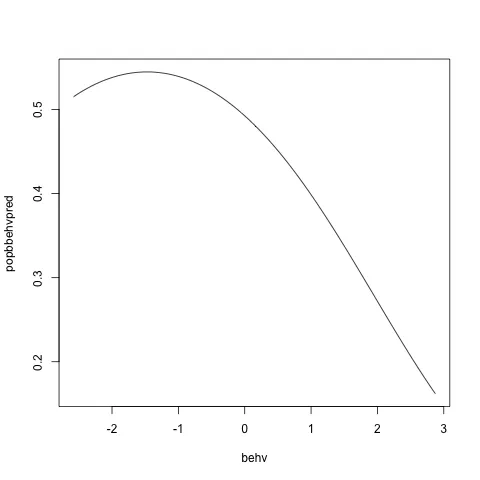
尽管我的虚假数据中没有任何显著性,但我们可以看到对于B群体,行为值较高似乎导致交配减少。