我已经编写了下面的程序,以执行以下操作:
我已经添加了代码,以便在数据/变量不再有用时立即清除内存。我也尽快关闭池。但是,即使使用14 GB的输入,我只期望2*14 GB的内存负担,但似乎发生了很多事情。我还尝试使用
我认为需要改进此代码位置,当我启动
- 将大型文本文件作为
pandas dataframe读取 - 然后使用特定列值进行
groupby将数据拆分并存储为数据帧列表。 - 然后将数据传输到
multiprocess Pool.map()中,以并行处理每个数据帧。
我已经添加了代码,以便在数据/变量不再有用时立即清除内存。我也尽快关闭池。但是,即使使用14 GB的输入,我只期望2*14 GB的内存负担,但似乎发生了很多事情。我还尝试使用
chunkSize 和 maxTaskPerChild 等调整,但在测试和大文件中都没有看到任何优化差异。我认为需要改进此代码位置,当我启动
multiprocessing 时。
p = Pool(3) # number of pool to run at once; default at 1
result = p.map(matrix_to_vcf, list(gen_matrix_df_list.values()))
但是,我会贴出整个代码。
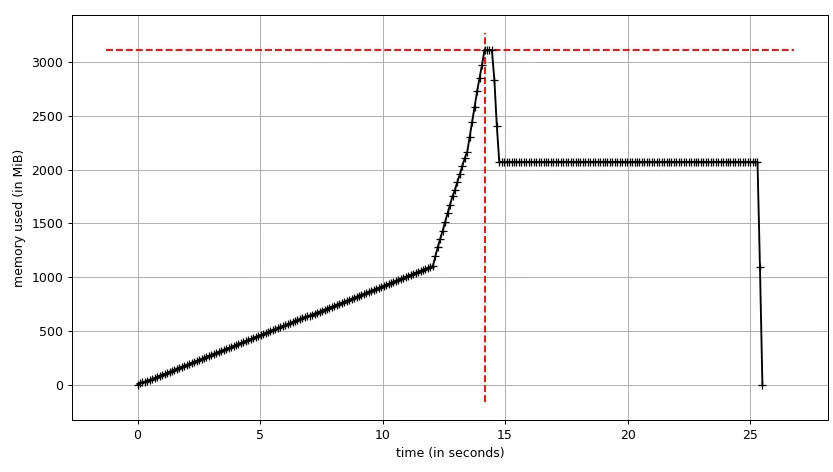
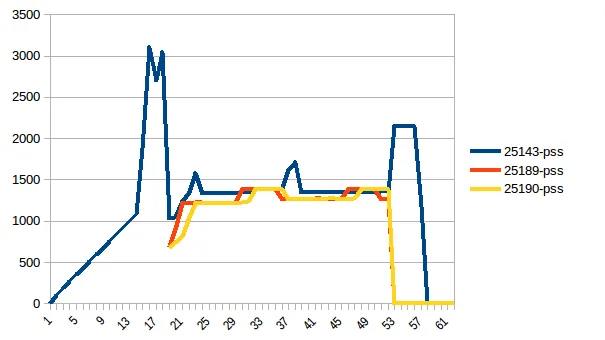
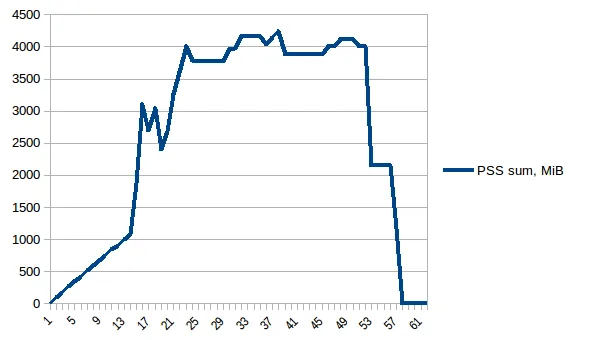
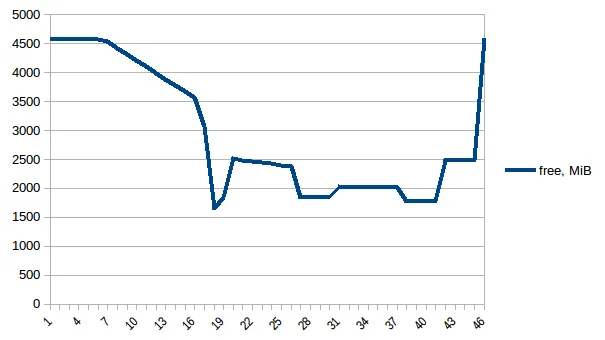
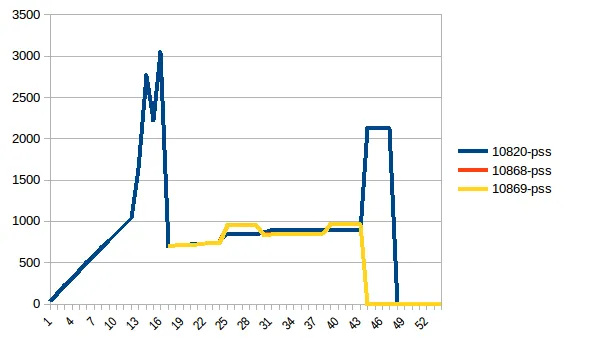
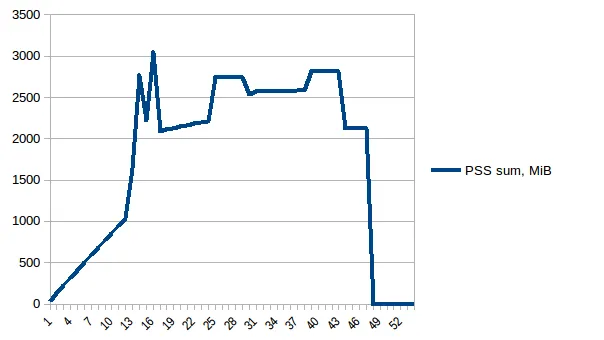
测试示例:我创建了一个测试文件("genome_matrix_final-chr1234-1mb.txt")最多250 mb并运行该程序。当我检查系统监视器时,可以看到内存消耗增加了约6 GB。我不是很清楚为什么250 mb文件加一些输出需要这么多内存空间。如果有助于查看真正的问题,我已通过Dropbox共享了该文件。https://www.dropbox.com/sh/coihujii38t5prd/AABDXv8ACGIYczeMtzKBo0eea?dl=0
有人能建议我如何解决这个问题吗?
我的Python脚本:
#!/home/bin/python3
import pandas as pd
import collections
from multiprocessing import Pool
import io
import time
import resource
print()
print('Checking required modules')
print()
''' change this input file name and/or path as need be '''
genome_matrix_file = "genome_matrix_final-chr1n2-2mb.txt" # test file 01
genome_matrix_file = "genome_matrix_final-chr1234-1mb.txt" # test file 02
#genome_matrix_file = "genome_matrix_final.txt" # large file
def main():
with open("genome_matrix_header.txt") as header:
header = header.read().rstrip('\n').split('\t')
print()
time01 = time.time()
print('starting time: ', time01)
'''load the genome matrix file onto pandas as dataframe.
This makes is more easy for multiprocessing'''
gen_matrix_df = pd.read_csv(genome_matrix_file, sep='\t', names=header)
# now, group the dataframe by chromosome/contig - so it can be multiprocessed
gen_matrix_df = gen_matrix_df.groupby('CHROM')
# store the splitted dataframes as list of key, values(pandas dataframe) pairs
# this list of dataframe will be used while multiprocessing
gen_matrix_df_list = collections.OrderedDict()
for chr_, data in gen_matrix_df:
gen_matrix_df_list[chr_] = data
# clear memory
del gen_matrix_df
'''Now, pipe each dataframe from the list using map.Pool() '''
p = Pool(3) # number of pool to run at once; default at 1
result = p.map(matrix_to_vcf, list(gen_matrix_df_list.values()))
del gen_matrix_df_list # clear memory
p.close()
p.join()
# concat the results from pool.map() and write it to a file
result_merged = pd.concat(result)
del result # clear memory
pd.DataFrame.to_csv(result_merged, "matrix_to_haplotype-chr1n2.txt", sep='\t', header=True, index=False)
print()
print('completed all process in "%s" sec. ' % (time.time() - time01))
print('Global maximum memory usage: %.2f (mb)' % current_mem_usage())
print()
'''function to convert the dataframe from genome matrix to desired output '''
def matrix_to_vcf(matrix_df):
print()
time02 = time.time()
# index position of the samples in genome matrix file
sample_idx = [{'10a': 33, '10b': 18}, {'13a': 3, '13b': 19},
{'14a': 20, '14b': 4}, {'16a': 5, '16b': 21},
{'17a': 6, '17b': 22}, {'23a': 7, '23b': 23},
{'24a': 8, '24b': 24}, {'25a': 25, '25b': 9},
{'26a': 10, '26b': 26}, {'34a': 11, '34b': 27},
{'35a': 12, '35b': 28}, {'37a': 13, '37b': 29},
{'38a': 14, '38b': 30}, {'3a': 31, '3b': 15},
{'8a': 32, '8b': 17}]
# sample index stored as ordered dictionary
sample_idx_ord_list = []
for ids in sample_idx:
ids = collections.OrderedDict(sorted(ids.items()))
sample_idx_ord_list.append(ids)
# for haplotype file
header = ['contig', 'pos', 'ref', 'alt']
# adding some suffixes "PI" to available sample names
for item in sample_idx_ord_list:
ks_update = ''
for ks in item.keys():
ks_update += ks
header.append(ks_update+'_PI')
header.append(ks_update+'_PG_al')
#final variable store the haplotype data
# write the header lines first
haplotype_output = '\t'.join(header) + '\n'
# to store the value of parsed the line and update the "PI", "PG" value for each sample
updated_line = ''
# read the piped in data back to text like file
matrix_df = pd.DataFrame.to_csv(matrix_df, sep='\t', index=False)
matrix_df = matrix_df.rstrip('\n').split('\n')
for line in matrix_df:
if line.startswith('CHROM'):
continue
line_split = line.split('\t')
chr_ = line_split[0]
ref = line_split[2]
alt = list(set(line_split[3:]))
# remove the alleles "N" missing and "ref" from the alt-alleles
alt_up = list(filter(lambda x: x!='N' and x!=ref, alt))
# if no alt alleles are found, just continue
# - i.e : don't write that line in output file
if len(alt_up) == 0:
continue
#print('\nMining data for chromosome/contig "%s" ' %(chr_ ))
#so, we have data for CHR, POS, REF, ALT so far
# now, we mine phased genotype for each sample pair (as "PG_al", and also add "PI" tag)
sample_data_for_vcf = []
for ids in sample_idx_ord_list:
sample_data = []
for key, val in ids.items():
sample_value = line_split[val]
sample_data.append(sample_value)
# now, update the phased state for each sample
# also replacing the missing allele i.e "N" and "-" with ref-allele
sample_data = ('|'.join(sample_data)).replace('N', ref).replace('-', ref)
sample_data_for_vcf.append(str(chr_))
sample_data_for_vcf.append(sample_data)
# add data for all the samples in that line, append it with former columns (chrom, pos ..) ..
# and .. write it to final haplotype file
sample_data_for_vcf = '\t'.join(sample_data_for_vcf)
updated_line = '\t'.join(line_split[0:3]) + '\t' + ','.join(alt_up) + \
'\t' + sample_data_for_vcf + '\n'
haplotype_output += updated_line
del matrix_df # clear memory
print('completed haplotype preparation for chromosome/contig "%s" '
'in "%s" sec. ' %(chr_, time.time()-time02))
print('\tWorker maximum memory usage: %.2f (mb)' %(current_mem_usage()))
# return the data back to the pool
return pd.read_csv(io.StringIO(haplotype_output), sep='\t')
''' to monitor memory '''
def current_mem_usage():
return resource.getrusage(resource.RUSAGE_SELF).ru_maxrss / 1024.
if __name__ == '__main__':
main()
赏金猎人更新:
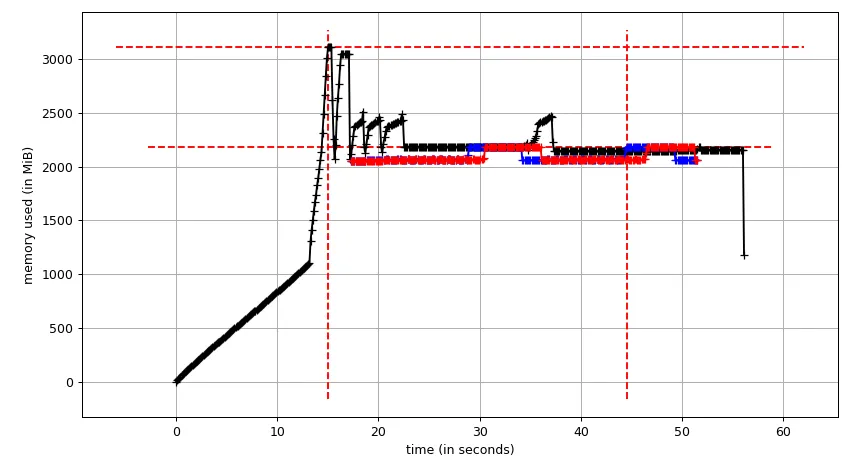
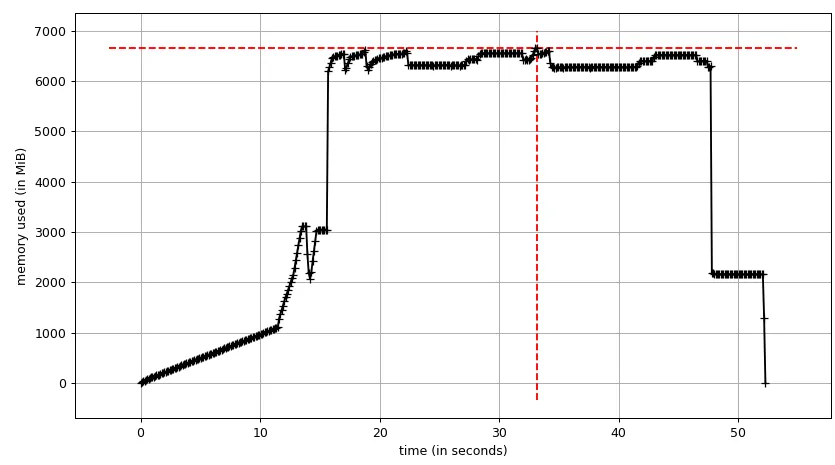
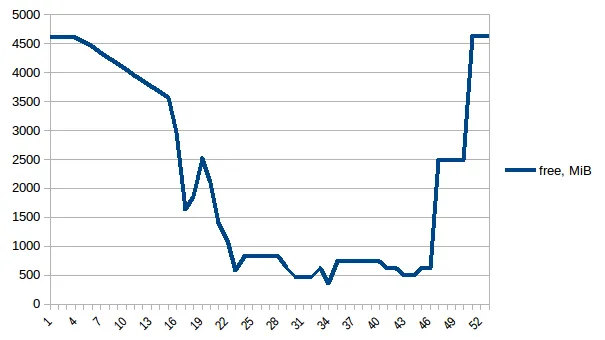
我已经使用Pool.map()实现了多进程,但是代码会导致巨大的内存负担(输入测试文件约300 MB,但内存负担约为6 GB)。我只期望最多有3 * 300 MB的内存负担。
- 有人能解释一下,对于这样一个小文件和如此短的计算长度,是什么导致了如此巨大的内存需求吗?
- 另外,我正在尝试采取答案并将其用于改进我的大型程序中的多进程。因此,添加任何方法、模块都不应该太大幅度地改变计算部分(CPU绑定进程)的结构。
- 我已经包含了两个测试文件以进行测试。
- 附加的代码是完整的,所以当复制粘贴时应该按预期工作。任何更改都应该仅用于改进多进程步骤的优化。