由于您没有适应函数,您需要将遗传算法视为分类器。因此,您需要想出一种评估单个染色体的方法。正如其他人建议您的那样,这是一个纯粹的分类问题,而不是优化问题,但是如果您仍然想使用遗传算法,下面是一些尝试初步方法的步骤:
您需要:
一个有效染色体的描述(如何编码)
要使用遗传算法,所有解决方案必须具有相同的长度(还有更高级的可变长度编码方法,但我不会涉及到那里)。因此,有了这一点,您需要找到一种最佳的编码方法。知道您的输入是一个可变长度的字符串,您可以将染色体编码为字母表的查找表(在Python中为字典)。但是,当您尝试应用交叉或突变操作时,字典会给您带来一些问题,因此最好将字母表和染色体编码拆分。参考语言模型,您可以检查n-grams,并且您的染色体将具有与字母表长度相同的长度:
.. Unigrams
alphabet = "ABCDE"
chromosome1 = [1, 2, 3, 4, 5]
chromosome2 = [1, 1, 2, 1, 0]
.. 二元组
alphabet = ["AB", "AC", "AD", "AE", "BC", "BD", "BE", "CD", "CE", "DE"]
chromosome = [1, 2, 3, 4, 5, 6, 7, 8, 9, 10]
.. 三元组
alphabet = ["ABC", "ABD", "ABE"...]
chromosome = as above, a value for each combination
2. 解码染色体以评估单个输入
您的染色体将为字母表中的每个元素表示一个整数值。因此,如果您想知道具有染色体的一个输入(可变长度字符串)的值,您将需要尝试一些评估函数,最简单的是每个字母值的总和。
alphabet = "ABC"
chromosome = [1, 2, 1]
input = "ABBBC"
value = reduce(lambda acc, x: acc + chromosme[alphabet.index(x)], input, 0)
3. 适应函数
你的适应函数只是一个简单的误差函数。你可以使用简单的误差和,平方误差... 单个基因的简单评估函数:
def fitnessFunction(inputs, results, alphabet, chromosome):
error = 0
for i in range(len(inputs)):
value = reduce(lambda acc, x: acc + chromosome[alphabet.index(x)], inputs[i], 0)
diff = abs(results[i] - value)
error += diff
return error
fitnessFunction(["ABC", "ABB", "ABBC"], [1,2,3], "ABC", [1, 1, 0])
现在,你可以使用任何交叉和变异运算符(例如:单点交叉和位翻转变异),找到使误差最小的染色体。
以下是改善算法模型的尝试:
- 使用bigrams或trigrams
- 更改评估方法(当前是查找表值的总和,可以是乘积或更复杂的东西)
- 尝试在染色体中使用实数而不仅仅是整数
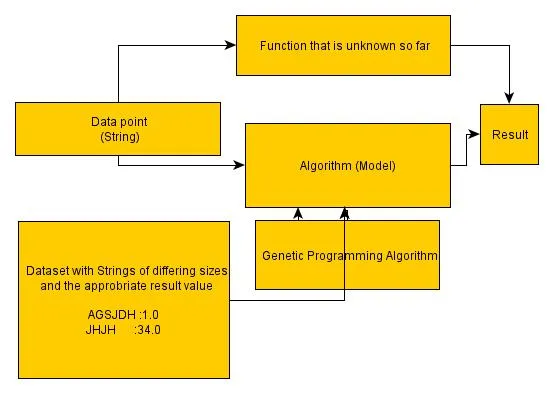 我想做的就是输入一个数据点并将其放入函数中。 然后我得到一个值,这是我的结果。不幸的是,我不知道这个函数,我只有一些示例数据集(可能只有1000个示例)。现在我使用遗传编程算法来找到一个算法,使其能够将我的数据点转换为结果。 这就是我的模型。 在这种情况下我遇到的问题是,数据点的长度不同。 对于固定长度,我可以将字符串中的每个字符指定为输入参数。但是如果我有不同数量的输入参数该怎么办呢?
我想做的就是输入一个数据点并将其放入函数中。 然后我得到一个值,这是我的结果。不幸的是,我不知道这个函数,我只有一些示例数据集(可能只有1000个示例)。现在我使用遗传编程算法来找到一个算法,使其能够将我的数据点转换为结果。 这就是我的模型。 在这种情况下我遇到的问题是,数据点的长度不同。 对于固定长度,我可以将字符串中的每个字符指定为输入参数。但是如果我有不同数量的输入参数该怎么办呢?