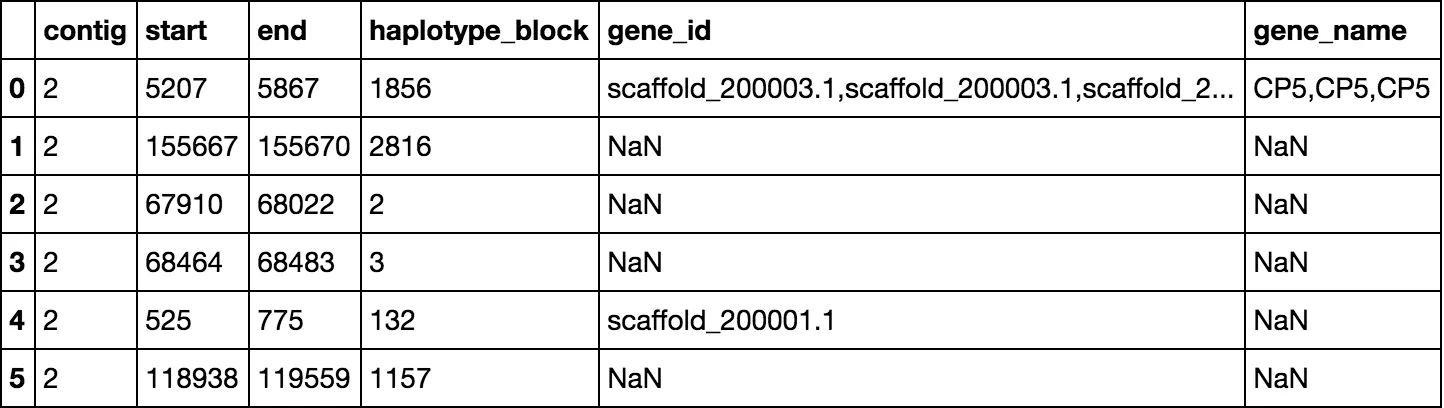
在以下数据中:
data01 =
contig start end haplotype_block
2 5207 5867 1856
2 155667 155670 2816
2 67910 68022 2
2 68464 68483 3
2 525 775 132
2 118938 119559 1157
data02 =
contig start last feature gene_id gene_name transcript_id
2 5262 5496 exon scaffold_200003.1 CP5 scaffold_200003.1
2 5579 5750 exon scaffold_200003.1 CP5 scaffold_200003.1
2 5856 6032 exon scaffold_200003.1 CP5 scaffold_200003.1
2 6115 6198 exon scaffold_200003.1 CP5 scaffold_200003.1
2 916 1201 exon scaffold_200001.1 NA scaffold_200001.1
2 614 789 exon scaffold_200001.1 NA scaffold_200001.1
2 171 435 exon scaffold_200001.1 NA scaffold_200001.1
2 2677 2806 exon scaffold_200002.1 NA scaffold_200002.1
2 2899 3125 exon scaffold_200002.1 NA scaffold_200002.1
问题:
- 我想比较这两个数据框的范围(起始-结束)。
- 如果范围重叠,我想要将data02中的
gene_id和gene_name值转移到data01的新列中。
我尝试了以下方法(使用pandas):
data01['gene_id'] = ""
data01['gene_name'] = ""
data01['gene_id'] = data01['gene_id'].\
apply(lambda x: data02['gene_id']\
if range(data01['start'], data01['end'])\
<= range(data02['start'], data02['last']) else 'NA')
如何改进这段代码?我目前使用的是pandas,但如果使用字典能更好地解决问题,我也愿意尝试。但请解释一下过程,我希望学习而不仅仅是得到答案。
谢谢,
期望输出:
contig start end haplotype_block gene_id gene_name
2 5207 5867 1856 scaffold_200003.1,scaffold_200003.1,scaffold_200003.1 CP5,CP5,CP5
# the gene_id and gene_name are repeated 3 times because three intervals (i.e 5262-5496, 5579-5750, 5856-6032) from data02 overlap(or touch) the interval ranges from data01 (5207-5867)
# So, whenever there is overlap of the ranges between two dataframe, copy the gene_id and gene_name.
# and simply NA on gene_id and gene_name for non overlapping ranges
2 155667 155670 2816 NA NA
2 67910 68022 2 NA NA
2 68464 68483 3 NA NA
2 525 775 132 scaffold_200001.1 NA
2 118938 119559 1157 NA NA