我正在与UpSetR进行比较,我想保存落入每个交集的元素列表。这可能吗?我无法在任何地方找到它...
手动完成这项任务将非常繁琐(由于有许多列表),而且由于它们已经被计算,不能保存它们感到令人沮丧。
手动完成这项任务将非常繁琐(由于有许多列表),而且由于它们已经被计算,不能保存它们感到令人沮丧。
str(list_filter)
List of 9
$ CellAge_Induces : chr [1:153] "AAK1" "ABI3" "ADCK5" "AGT" ...
$ CellAge_Inhibits : chr [1:121] "ACLY" "AKR1B1" "ASPH" "ATF7IP" ...
$ CLASSICAL_SASP : chr [1:38] "BGN" "CCL2" "CCL20" "COL1A1" ...
$ FRIDMAN_SENESCENCE_UP : chr [1:77] "ALDH1A3" "CCND1" "CD44" "CDKN1A" ...
$ ISM_SCORE : chr [1:128] "HSH2D" "OTOF" "TRIM69" "PSME1" ...
$ MOSERLE_IFNA_RESPONSE : chr [1:31] "CD274" "CMPK2" "CXCL10" "DDX58" ...
$ REACTOME_SENESCENCE_SASP: chr [1:110] "ANAPC1" "ANAPC10" "ANAPC11" "ANAPC15" ...
$ SAEPHIA_CURATED_SASP : chr [1:38] "IL1A" "IL1B" "CXCL10" "CXCL1" ...
$ senmayo : chr [1:125] "ACVR1B" "ANG" "ANGPT1" "ANGPTL4" ...
df2 <- data.frame(gene=unique(unlist(list_filter)))
head(df2)
gene
1 AAK1
2 ABI3
3 ADCK5
4 AGT
5 AKT1
6 ALOX15B
dim(df2)
[1] 671 1
其中一个是列表的“数据框架”版本。包含签名中的每个基因以及每个签名(集合)的名称。
df1 <- lapply(list_filter,function(x){
data.frame(gene = x)
}) %>%
bind_rows(.id = "path")
head(df1)
path gene
1 CellAge_Induces AAK1
2 CellAge_Induces ABI3
3 CellAge_Induces ADCK5
4 CellAge_Induces AGT
5 CellAge_Induces AKT1
6 CellAge_Induces ALOX15B
dim(df1)
[1] 821 2
df_int <- lapply(df2$gene,function(x){
# pull the name of the intersections
intersection <- df1 %>%
dplyr::filter(gene==x) %>%
arrange(path) %>%
pull("path") %>%
paste0(collapse = "|")
# build the dataframe
data.frame(gene = x,int = intersection)
}) %>%
bind_rows()
head(df_int,n=20)
gene int
1 AAK1 CellAge_Induces
2 ABI3 CellAge_Induces
3 ADCK5 CellAge_Induces
4 AGT CellAge_Induces
5 AKT1 CellAge_Induces
6 ALOX15B CellAge_Induces
7 AR CellAge_Induces
8 ARPC1B CellAge_Induces
9 ASF1A CellAge_Induces
10 AXL CellAge_Induces|senmayo
11 BHLHE40 CellAge_Induces
12 BLK CellAge_Induces
13 BRAF CellAge_Induces
14 BRD7 CellAge_Induces
15 CAV1 CellAge_Induces
16 CCND1 CellAge_Induces|FRIDMAN_SENESCENCE_UP
17 CDK18 CellAge_Induces
18 CDKN1A CellAge_Induces|FRIDMAN_SENESCENCE_UP|REACTOME_SENESCENCE_SASP
19 CDKN1C CellAge_Induces|FRIDMAN_SENESCENCE_UP
20 CDKN1B CellAge_Induces|REACTOME_SENESCENCE_SASP
dim(df_int)
[1] 671 2
df_int %>%
group_by(int) %>%
summarise(n=n()) %>%
arrange(desc(n))
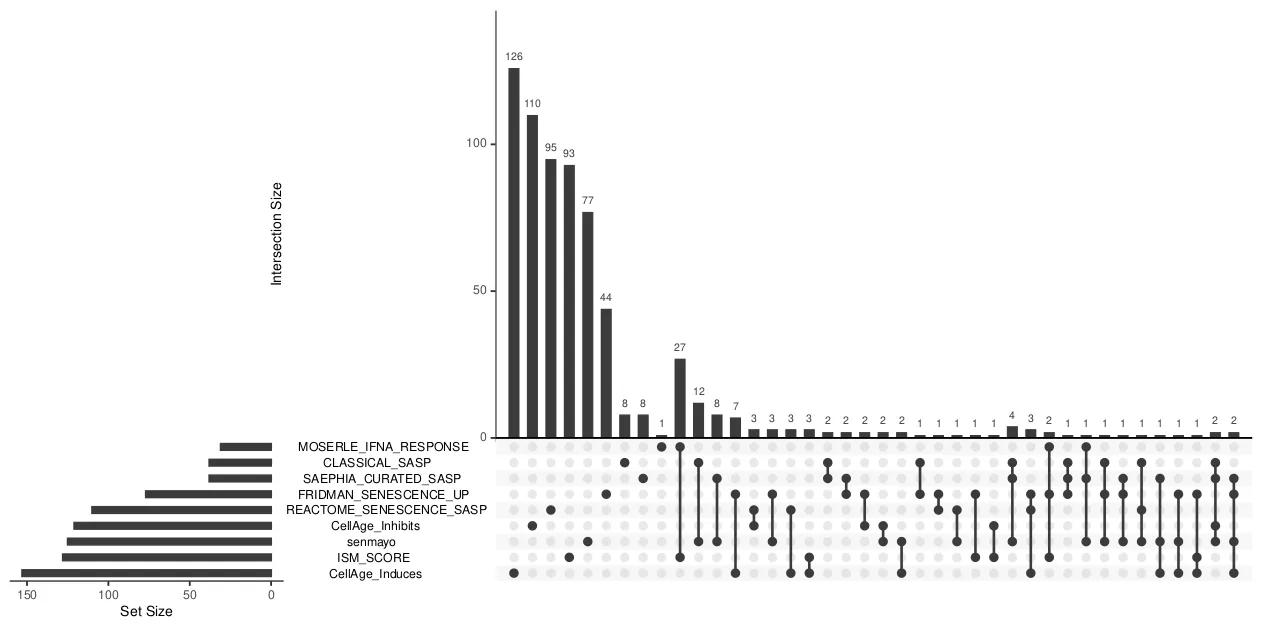
# A tibble: 47 × 2
int n
<chr> <int>
1 CellAge_Induces 126
2 CellAge_Inhibits 110
3 REACTOME_SENESCENCE_SASP 95
4 ISM_SCORE 93
5 senmayo 77
6 FRIDMAN_SENESCENCE_UP 44
7 ISM_SCORE|MOSERLE_IFNA_RESPONSE 27
8 CLASSICAL_SASP|senmayo 12
9 CLASSICAL_SASP 8
10 SAEPHIA_CURATED_SASP 8
# … with 37 more rows
# ℹ Use `print(n = ...)` to see more rows
upset(fromList(list_filter),nsets = 10)
data <- data.frame(
entry = paste0("Entry.", 1:10),
"A" = c(0,0,0,0,1,0,1,1,0,0),
"B" = c(1,0,0,0,1,1,1,1,1,0),
"C" = c(1,1,1,1,0,0,1,0,1,1)
)
# NOT REQUIRED. Only to confirm that upset works with these data
upset(data)
您可以通过将所有二进制列粘贴在一起来识别交叉点。我使用 unite 便捷函数实现此功能:
注意:根据您的数据是否具有行名称或带名称的列,您可能需要更改此设置。
data_with_intersection <- data %>%
unite(col = "intersection", -c("entry"), sep = "")
从这里开始,您可以轻松计算每个交集的大小:
# Table of intersections and the number of entries
data_with_intersection %>%
group_by(intersection) %>%
summarise(n = n()) %>%
arrange(desc(n))
甚至可以提取每个交集中条目/元素的列表:
# List of intersections and their entries
data_with_intersection %>%
group_by(intersection) %>%
summarise(list = list(entry)) %>%
mutate(list = setNames(list, intersection)) %>%
pull(list)
目前还没有现成的upSetR函数来处理这个(但是)。不过,可以提取它:
library(UpSetR)
# Example input as list, expected output is 1 and 5:
listInput <- list(one = c(1, 2, 3, 5, 7, 8, 11, 12, 13),
two = c(1, 2, 4, 5, 10),
three = c(1, 5, 6, 7, 8, 9, 10, 12, 13))
当分配的异常返回一个值时,该值还包括数据:
x <- upset(fromList(listInput))
x$New_data
# one two three
# 1 1 1 1
# 2 1 1 0
# 3 1 0 0
# 4 1 1 1
# 5 1 0 1
# 6 1 0 1
# 7 1 0 0
# 8 1 0 1
# 9 1 0 1
# 10 0 1 0
# 11 0 1 1
# 12 0 0 1
# 13 0 0 1
从这里我们可以看到第一行和第四行在所有三个集合中都存在。项目的顺序是基于它们在列表中出现的顺序定义的,如下所示:
x1 <- unlist(listInput, use.names = FALSE)
x1 <- x1[ !duplicated(x1) ]
x1
# [1] 1 2 3 5 7 8 11 12 13 4 10 6 9
x1[ rowSums(x$New_data) == 3 ]
# [1] 1 5
或者我们可以只使用Reduce:
Reduce(intersect, listInput)
# [1] 1 5
x <- upset(...),它会返回用于绘图的数据。我无法轻易地在那个 x 对象中找到你需要的信息。 - zx8754